
Current Projects
Overview
- Role of genetic ancestry in COVID-19 susceptibility and severity
- Identifying novel orphan genes in human using massive RNA-Seq data
- Examining cell-type specific expression of novel genes
- A deep-generative model to integrate massive bulk expression datasets
- MetaOmGraph: A tool for exploration of big omics data
- pyrpipe: Bioinformatics pipelines in pure-python
- orfipy: Fast and flexible ORF caller
- Past Projects
Role of genetic ancestry in COVID-19 susceptibility and severity
As of January 2021, the COVID-19 pandemic has infected over 98 million people and killed over 2 million worldwide. People of African and Latin ancestries in the U.S. are disproportionately affected by COVID-19 with respect to prevalence, morbidity, and mortality.
An individual’s health is a result of complex interactions among one’s genetic, environmental, and socio-economic factors. We leveraged RNA-Seq expression data and probed differences in gene-expression among people from European and African Ancestries. Although we could not control environmental and socio-economic factors (because of lack of metadata), we found differential expression of genes implicated in COVID-19 infection. Further, African American gene expression signatures reveal an increased number or activity of esophageal glandular cells and lung ACE2-positive basal keratinocytes. Please read our preprint here.
Our ongoing research, in collaboration with the COVID-19 International Research Team (COV-IRT), aims to detect genetic factors that can explain the disparity of COVID-19 infection and mortality rate among different populations. We are particularly interested in identifying and examining the roles of human specific orphan genes and non-coding RNAs in COVID-19 infection.
This research is funded by National Science Foundation grant IOS 1546858, Orphan Genes: An Untapped Genetic Reservoir of Novel Traits, and by the Center for Metabolic Biology, Iowa State University
Identifying novel orphan genes in human using massive RNA-Seq data
 Figure showing life cycle of orphan genes. Ref: Singh, U., & Wurtele, E. S. (2020). Genetic novelty: How new genes are born. Elife, 9, e55136.
Figure showing life cycle of orphan genes. Ref: Singh, U., & Wurtele, E. S. (2020). Genetic novelty: How new genes are born. Elife, 9, e55136.
During evolution, genes undergo duplication and divergence and give rise to new genes.
These new genes can potentially code for new proteins and provide the organism with useful biological functions.
Genes resulting through the process of duplication could be traced back to their ancestral genes via sequence homology.
However, in every organism, a number of genes can not be traced back to any ancestral gene.
These species-specific genes are termed orphan genes.
Many such orphan genes are involved in biologically important functions.
Orphan genes can arise de novo from non-genic regions in the genome.
The focus of my research is to identify and characterize novel expressed genes in humans. To achieve this goal, I am integrating and analyzing terabytes high quality of RNA-Seq datasets from massive projects like GTEx, TCGA and SRA.
This research is funded by National Science Foundation grant IOS 1546858, Orphan Genes: An Untapped Genetic Reservoir of Novel Traits, and by the Center for Metabolic Biology, Iowa State University
Examining cell-type specific expression of novel genes
With the advent of single-cell sequencing it is now easy to examine transcriptome profiles at the single-cell level. I am leading a small team to examine publicly available single-cell RNA-Seq datasets to look for cell-specific expression of novel genes.
This research is funded by National Science Foundation grant IOS 1546858, Orphan Genes: An Untapped Genetic Reservoir of Novel Traits, and by the Center for Metabolic Biology, Iowa State University
A deep-generative model to integrate massive bulk expression datasets
Petabytes of RNA-Seq are available publicly via resources like NCBI-SRA. Integrative analysis of such data provides an opportunity to mine biologically essential insights. A significant challenge of integrating RNA-Seq datasets from multiple sources is unwanted technical and biological effects. Technical effects, such as batch-effects due to technical factors, may be controlled if the batch co-variates are known. A significant problem arises when sources of such effects are unknown or hidden. Methods like SVA can remove some of the unwanted variations. New methods like SAUCIE use auto-encoders for batch correction and de-noising single-cell expression data, in an unsupervised manner. Other deep-learning-based methods have also been developed for single-cell datasets.
I am interested in developing deep generative models for non-linear factor analysis of massive bulk RNA-Seq expression data. In this ongoing project, I am using variational auto-encoders to model RNA-Seq count data directly and project them into smaller latent dimensions to remove unwanted variation.
This research is funded by National Science Foundation grant IOS 1546858, Orphan Genes: An Untapped Genetic Reservoir of Novel Traits, and by the Center for Metabolic Biology, Iowa State University
MetaOmGraph: A tool for exploration of big omics data
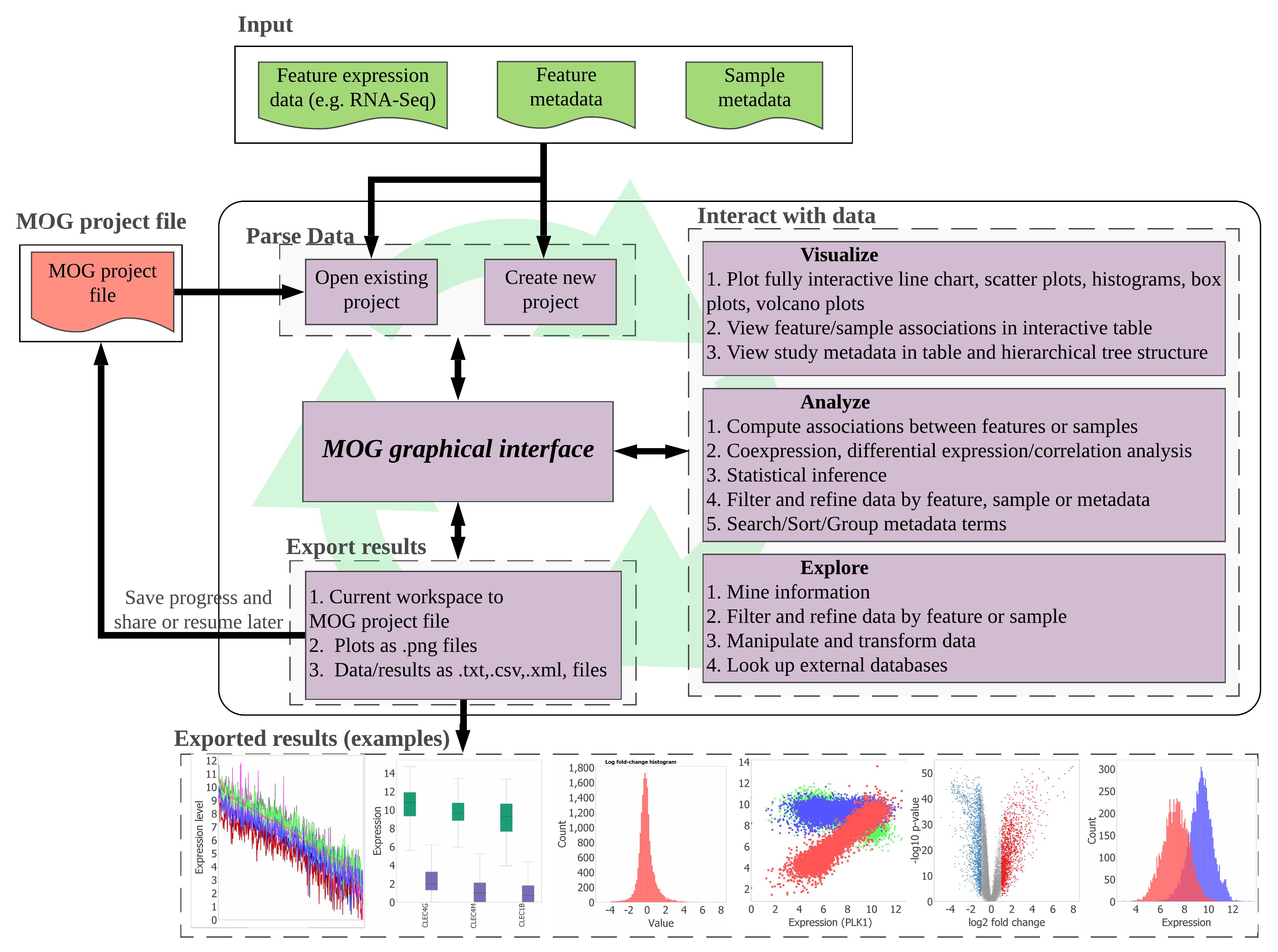 Figure showing overview of MetaOmGraph tool. Ref: Singh, U., Hur, M., Dorman, K., & Wurtele, E. S. (2020). MetaOmGraph: a workbench for interactive exploratory data analysis of large expression datasets. Nucleic acids research, 48(4), e23-e23.
Figure showing overview of MetaOmGraph tool. Ref: Singh, U., Hur, M., Dorman, K., & Wurtele, E. S. (2020). MetaOmGraph: a workbench for interactive exploratory data analysis of large expression datasets. Nucleic acids research, 48(4), e23-e23.
I am the lead developer of MetaOmGraph, a fully interactive exploratory data analysis software for big omics datasets. MetaOmGraph allows user to visualize and analyze their data using a number methods in a fully interactive manner. As data becomes complex, it is important to explore data from different perspectives. MetaOmGraph lets the user do that in an efficient and straightforward manner. Domain experts, such as biologist, cell biologists, virologists, and medical professionals can easily analyze the data without writing any code. MetaOmGraph is an open source project. Please see it on github here.
This research is funded by National Science Foundation grant IOS 1546858, Orphan Genes: An Untapped Genetic Reservoir of Novel Traits, and by the Center for Metabolic Biology, Iowa State University
pyrpipe: Bioinformatics pipelines in pure-python
 Figure showing overview of pyrpipe. Ref: Singh, U., Li, J., Seetharam, A., & Wurtele, E. S. (2020). pyrpipe: a python package for RNA-Seq workflows. bioRxiv.
Figure showing overview of pyrpipe. Ref: Singh, U., Li, J., Seetharam, A., & Wurtele, E. S. (2020). pyrpipe: a python package for RNA-Seq workflows. bioRxiv.
Writing complex bioinformatics pipelines in a fully reproducible, easy-to-debug, maintainable manner can be challenging. Often plain scripting is used to implement the pipelines, which can have many drawbacks. I developed pyrpipe to provide a framework to implement RNA-Seq and other bioinformatics pipelines in pure python in an object-oriented manner. pyrpipe provides special API classes that allow importing any Unix executable command in python. All commands executed via pyrpipe is in this manner is automatically logged, monitored, or can be flexibly controlled using pyrpipe options. pyrpipe comes with API to access popular RNA-Seq tools directly from python. Using this framework, RNA-Seq processing pipelines can be intuitively coded. pyrpipe pipelines could be easily extended to add new tools. Parameters can be easily managed in YAML files, which are automatically parsed by pyrpipe. pyrpipe_diagnostic can generate in-depth reports and logs for debugging and reproducibility. pyrpipe can be easily installed via conda or PyPI. pyrpipe source code is available on github. Please read the pre-print here.
This research is funded by National Science Foundation grant IOS 1546858, Orphan Genes: An Untapped Genetic Reservoir of Novel Traits, and by the Center for Metabolic Biology, Iowa State University
orfipy: Fast and flexible ORF caller
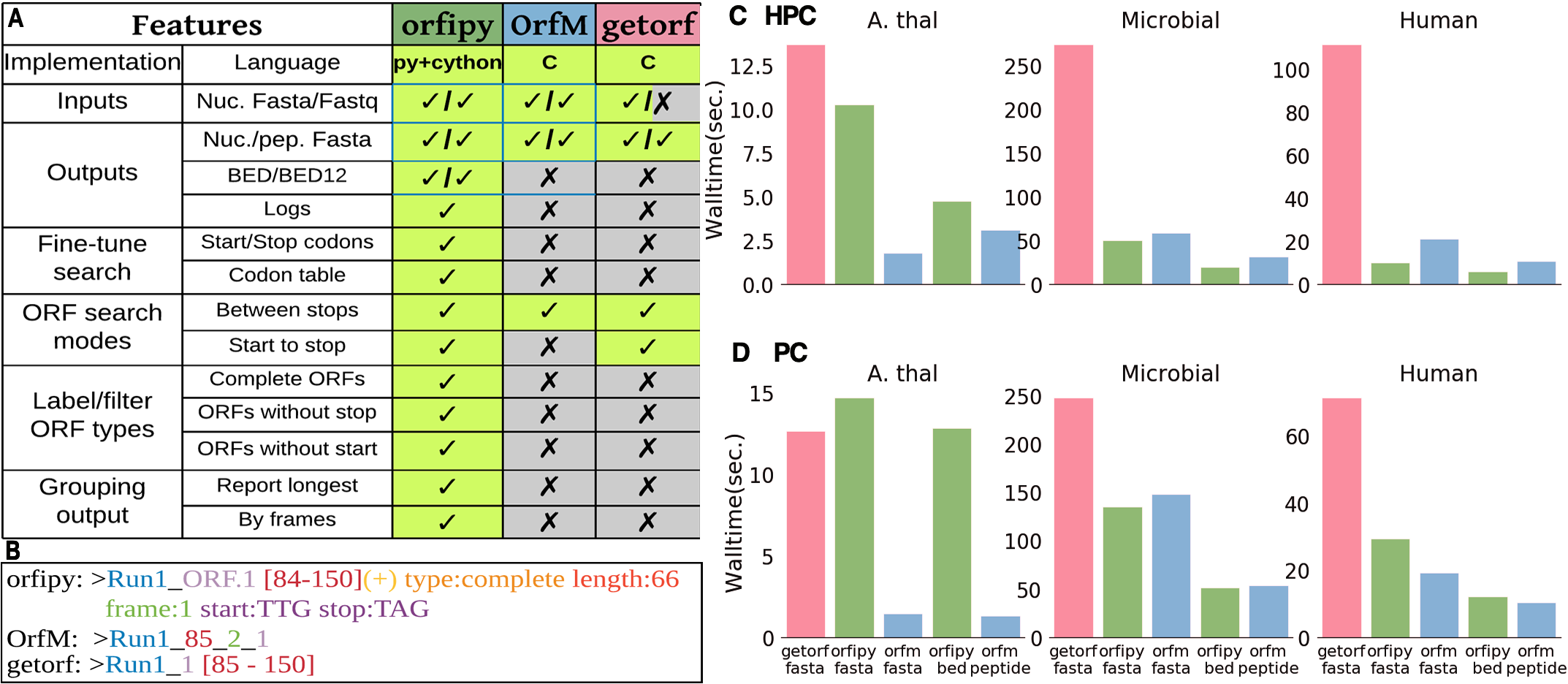 Figure showing features of orfipy. Ref: Singh, U., & Wurtele, E. S. (2020). orfipy: a fast and flexible tool for extracting ORFs. bioRxiv.
Figure showing features of orfipy. Ref: Singh, U., & Wurtele, E. S. (2020). orfipy: a fast and flexible tool for extracting ORFs. bioRxiv.
Identification of Open Reading Frames (ORF) is an integral step for any gene annotation pipeline. Presently, there exist many tools for ORF identification, such as the popular Getorf and OrfM. These tools work great but are not flexible in terms of fine-tuning ORF search. I developed orfipy to solve this challenge. orfipy is fast and flexible. orfipy reduced the total runtime of my orphan gene prediction pipeline significantly while accurately identifying the ORFs. orfipy can be easily installed via conda or PyPI. orfipy source code is available on github. Please read the pre-print here.
This research is funded by National Science Foundation grant IOS 1546858, Orphan Genes: An Untapped Genetic Reservoir of Novel Traits, and by the Center for Metabolic Biology, Iowa State University
Past Projects
coming soon…